Cribler rapidement des milliards de molécules pour trouver de nouveaux candidat-médicaments
Des scientifiques du Laboratoire d’innovation thérapeutique ont développé une méthode utilisant l'intelligence artificielle (SpaceDock) pour concevoir à la demande de nouveaux candidat-médicaments parmi des milliards de molécules possibles.
La recherche de candidat-médicaments repose essentiellement sur le criblage expérimental ou numérique du plus grand nombre possible de molécules dont on pense qu’elles pourraient présenter des vertus thérapeutiques. Pour un criblage expérimental, c’est le nombre de molécules disponibles pour les essais biologiques qui détermine le périmètre du criblage, périmètre que l’on peut étendre à d’autres systèmes chimiques « théoriques » par un criblage virtuel.
Le nombre et les performances thérapeutiques des molécules pouvant passer avec succès les tests biologiques dépendent essentiellement de l’« espace chimique » criblé : plus la banque de molécules est importante en terme de taille et de diversité chimique, meilleure sera l’efficacité du criblage.
Des scientifiques du Laboratoire d'innovation thérapeutique (CNRS-Université de Strasbourg) viennent de concevoir des inhibiteurs particulièrement efficaces de protéines d'intérêt thérapeutique en criblant virtuellement des espaces chimiques théoriques ultra-larges (jusqu'à 5 milliards de molécules). La méthode de criblage qu’ils ont mise au point leur a permis de cibler seulement quinze candidat-médicaments dont ils ont réalisé la synthèse, puis l'évaluation biologique.
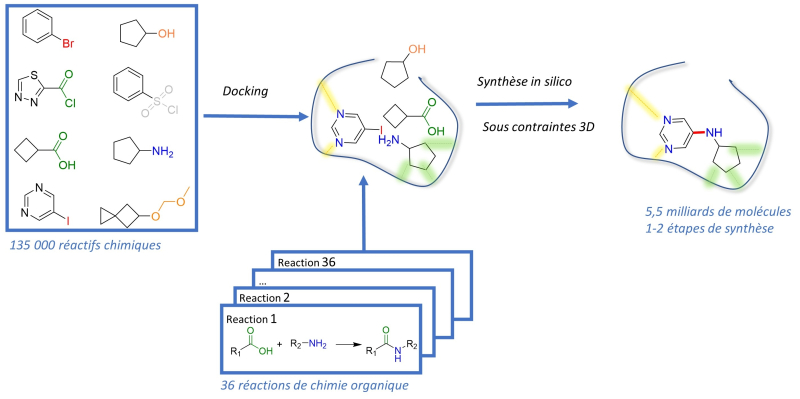
Appelée "SpaceDock", cette approche innovante débute par l’identification et la localisation des sites actifs de la protéine avec lesquels les inhibiteurs pourraient interagir. Pour le récepteur D3 à la dopamine, 135 000 réactifs chimiques commerciaux susceptibles de s’ancrer à ce site actif ont ainsi pu être ainsi sélectionnés. Les scientifiques ont ensuite pris en compte les contraintes topologiques de la structure tridimensionnelle de la protéine, leur indiquant parmi ces réactifs ceux qui vont pouvoir se retrouver suffisamment proches pour interagir et former de nouvelles molécules… Des centaines de milliards de rapprochements sont ainsi possibles dont on peut prédire le devenir en suivant les modes opératoires de 40 réactions « classiques » de chimie organique. La banque de criblage la plus importante jamais constituée…
Ultra-rapide, sélective et fiable, cette méthode, publiée dans la revue ACS Cent. Sci., permet de prédire, synthétiser, puis tester des molécules à vocation thérapeutique en puisant dans une palette de substances chimiques inédite par sa taille, sa pertinence et sa diversité
Rédacteur : CCdM
Référence
François Sindt, Anthony Seyller, Merveille Eguida & Didier Rognan
Protein Structure-Based Organic Chemistry-Driven Ligand Design from Ultralarge Chemical Spaces
ACS Cent. Sci. 2024